An rising quantity of research contain integrative evaluation of gene and protein expression knowledge, taking benefit of new applied sciences equivalent to next-generation transcriptome sequencing and extremely delicate mass spectrometry (MS) instrumentation. Recently, a technique, termed ribosome profiling (or RIBO-seq), based on deep sequencing of ribosome-protected mRNA fragments, not directly monitoring protein synthesis, has been described.
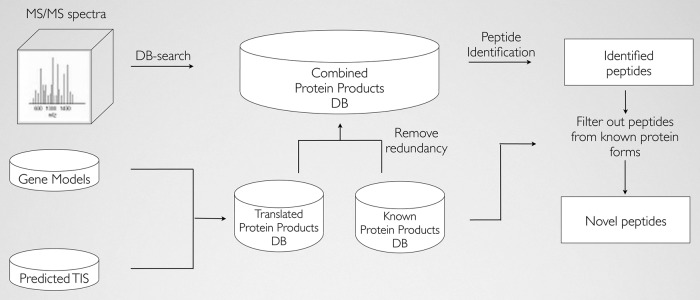
We devised a proteogenomic strategy establishing a {custom} protein sequence search house, constructed from each Swiss-Prot- and RIBO-seq-derived translation products, relevant for MS/MS spectrum identification. To file the impression of utilizing the constructed deep proteome database, we carried out two alternative MS-based proteomic methods as follows: (i) a daily shotgun proteomic and (ii) an N-terminal mixed fractional diagonal chromatography (COFRADIC) strategy. Although the previous method provides an general evaluation on the protein and peptide degree, the latter method, particularly enabling the isolation of N-terminal peptides, could be very applicable in validating the RIBO-seq-derived (alternative) translation initiation website profile.
We exhibit that this proteogenomic strategy will increase the general protein identification price 2.5% (e.g. new protein products, new protein splice variants, single nucleotide polymorphism variant proteins, and N-terminally prolonged kinds of recognized proteins) as in contrast with solely looking UniProtKB-SwissProt. Furthermore, utilizing this practice database, identification of N-terminal COFRADIC knowledge resulted in detection of 16 alternative begin websites giving rise to N-terminally prolonged protein variants apart from the identification of 4 translated upstream ORFs. Notably, the characterization of these new translation products revealed the use of a number of near-cognate (non-AUG) begin codons.
As deep sequencing methods have gotten extra commonplace, inexpensive, and widespread, we anticipate that mRNA sequencing and particularly custom-tailored RIBO-seq will turn into indispensable within the MS-based protein or peptide identification course of. The underlying mass spectrometry proteomics knowledge have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD000124.
Nuclear receptors and coregulators are multifaceted gamers in regular metabolic and homeostatic processes along with a range of illness states together with most cancers, irritation, diabetes, weight problems, and atherosclerosis.
Over the previous 7 yr, the Nuclear Receptor Signaling Atlas (NURSA) analysis consortium has labored towards establishing a discovery-driven platform designed to deal with key questions regarding the expression, group, and operate of these molecules in a range of experimental mannequin methods. By making use of highly effective applied sciences equivalent to quantitative PCR, high-throughput mass spectrometry, and embryonic stem cell manipulation, we’re pursuing these questions in a collection of transcriptomics-, proteomics-, and metabolomics-based analysis initiatives and assets.
The consortium‘s site (www.nursa.org) integrates NURSA datasets and current public datasets with the final word aim of furnishing the bench scientist with a complete framework for speculation technology, modeling, and testing. We place a robust emphasis on neighborhood enter into the event of this useful resource and to this finish have printed datasets from educational and industrial laboratories, established strategic alliances with Endocrine Society journals, and are growing instruments to permit site customers to behave as knowledge curators. With the continued help of the nuclear receptor and coregulator signaling communities, we imagine that NURSA could make an enduring contribution to analysis on this dynamic area.
UPDATES IN PROTEOMECENTRAL
Once an information set turns into publicly obtainable (normally when the related journal article is printed), the receiving repository transmits a message coded in PX XML to the ProteomeCentral server. All metadata within the PX XML message are registered and made obtainable within the publicly accessible ProteomeCentral portal . Since the unique model 1.0, the PX XML schema has been up to date to model 1.3 (accessible at to accommodate further data associated to laboratory heads chargeable for knowledge units as a long run contact level than the submitter and help the operations of the brand new PX members. The precise submitted knowledge set information stay within the dealing with repository, however are linked from ProteomeCentral. Thus, PX implements a big scale, distributed database infrastructure. Users can question all PX public knowledge units utilizing completely different organic and technical metadata, key phrases, tags or references, irrespective of which unique PX dealing with repository shops the information set information. It is necessary to spotlight that password-protected knowledge units (present process the assessment course of) are solely accessible within the unique repository, though there are directions for reviewers on entry them at ProteomeCentral.
The ProteomeCentral consumer interface has additionally been up to date to include improved abstract and exploratory visualizations. There are actually graphical widgets that enable customers to visualise the relative proportions of knowledge units throughout all repositories by species and by instrument mannequin, in addition to phrase clouds that depict probably the most generally repeated title phrases and knowledge set-associated key phrases. As the consumer clicks on these visualizations, the tabular itemizing of knowledge units immediately updates to mirror the picks.
OVERALL DATA SUBMISSION AND DATA ACCESS STATISTICS
Figure 2 reveals an aggregated and abstract view of PX knowledge gathered since 2012. By the top of July 2016, a complete of 4534 PX knowledge units had been submitted to any of the PX assets. In phrases of particular person assets, round 4067 knowledge units (representing 89.7% of all the information units), had been submitted to PRIDE, adopted by MassIVE (339 knowledge units), PASSEL (115 knowledge units) and jPOST (13 knowledge units, simply joined PX at the start of July 2016). Data units come from 50 nations, demonstrating the worldwide attain of the consortium. The most represented nations are USA (1105 knowledge units), Germany (546), United Kingdom (411), China (356) and France (229). Since 2012, the quantity of submitted knowledge units has elevated considerably yearly, starting from 102 (2012) to 1758 (2015). In the primary seven months of 2016, a complete of 1184 knowledge units had already been submitted.Figure 2.Summary of the primary metrics of ProteomeXchange submitted knowledge units (by the top of July 2016). The quantity of knowledge units is indicated for every PX useful resource, knowledge entry standing and for the highest species and nations represented. Re-analysed knowledge units usually are not included within the metrics.
Open knowledge can solely be open whether it is made public in a well timed method. But, very often, there’s not a easy strategy to join the information set to the printed papers, because the authors neglect to speak that the corresponding papers to submitted knowledge units have been printed. In the case of PX assets, 57% of these knowledge units (2597) have been publicly obtainable by July 2016, largely because of curation efforts to seek out papers related to submitted knowledge units. It is necessary to spotlight that the associate PX assets will make knowledge public, with out asking the unique submitter, as quickly as they discover out that the related paper has been printed.
Overall, knowledge units in PX come from 936 completely different taxonomy IDs (Supplementary Table S1). In most instances, the data supplied by the submitters is on the species degree. However, phrases like ‘intestine microbiome’ or ‘uncultured marine microorganism‘ could be chosen as properly as a substitute of itemizing the precise species. The prime 5 species (represented by greater than a 100 knowledge units) are: Homo sapiens (2010 knowledge units), Mus musculus (604), Saccharomyces cerevisiae (191), Arabidopsis thaliana (140) and Rattus norvegicus (127). Human samples (tissues, fluids or derived cell traces) signify roughly half of all of the PX knowledge units.
We additionally wish to spotlight that PXD accession numbers are actually extensively utilized in journal articles. PXD identifiers could be searched (utilizing the important thing ‘PXD00*’) in literature assets equivalent to PubMed, PubMedCentral and EuropePubMedCentral. As of September 2016, the numbers of publications referring to PX knowledge units have been 558, 548 and 1426, respectively. The purpose for the distinction is that PubMed searches embody solely the textual content of the abstracts, whereas the opposite two assets search the complete textual content of the articles.
PROTEOMEXCHANGE AND THE HUMAN PROTEOME PROJECT DATA GUIDELINES
The Human Proteome Project (HPP) is a global effort by the Human Proteome Organization to increase our understanding of your complete complement of proteins encoded by the human genome. In 2012, the HPP launched model 1.0 of its knowledge tips through which all knowledge printed in help of the HPP was required to be submitted to a PX repository. This was the primary main entity particularly to make PX deposition obligatory. In a just lately revised and expanded set of model 2.1 tips , PX deposition continues to be obligatory, now on the ‘full’ submission degree. This HPP deposition requirement has vastly contributed to rising the information deposition charges of human proteome associated knowledge locally as an entire.
DISCUSSION AND FUTURE PLANS
In the previous few years, the PX consortium has established itself as a community-responsive set of dependable proteomics knowledge repositories to allow knowledge submission and dissemination of MS proteomics experiments. With help from funding companies and scientific journals, PX is actively altering the information sharing tradition within the area by selling and enabling sharing of proteomics knowledge within the public area. For instance, after assessing the steadiness of PX assets, since July 2015, the journal Molecular and Cellular Proteomics, one of probably the most outstanding proteomics scientific journals, is now once more mandating deposition of uncooked knowledge with each submitted manuscript , and different journals are transferring on this course (e.g. journals from the Nature and PLOS teams). In this context, public knowledge availability and sharing within the area are shortly turning into the norm, as demonstrated by the excessive knowledge submissions statistics.
Sustainability of publically accessible repositories is an important issue within the PX Consortium. Consider the impression of the closure of Tranche and Peptidome because of the cessation of NIH funding and the partial loss of each knowledge units and widespread public confidence had on the proteomics neighborhood. To guarantee long-term knowledge availability, the PX members have dedicated to import the information obtainable in some other of the PX assets if it has funding issues and should stop its operations (see PX Consortium settlement at , because it occurred prior to now regrettably for proteomics assets equivalent to Tranche (whose hundreds of surviving knowledge units have been imported into MassIVE) and Peptidome (whose knowledge units have been imported into PRIDE) , earlier than PX was formally began. As a further incentive towards sustainability, knowledge may also be reproduced throughout repositories to allow systematic reanalysis, as MassIVE presently illustrates with tens of terabytes in a whole bunch of knowledge units already imported from PRIDE for varied varieties of knowledge reanalysis.
In the long run, we are going to work on supporting ‘full’ submissions for different in style instruments and more and more in style knowledge workflows within the area, equivalent to DIA (MassIVE is already beginning to help some DIA knowledge workflows at current), prime down and MS imaging approaches. MaxQuant and ProteomeDiscoverer® are two extensively used instruments for DDA shotgun proteomics workflows that can’t be supported as ‘full’ submissions. This consortium will proceed to liaise with the builders of these instruments to resolve interface points and arrive at a seamless submission course of. In phrases of supporting different knowledge workflows, this can contain in parallel the event of appropriate open knowledge requirements.
It can also be anticipated that reanalysis of current knowledge units will solely improve. In addition to PeptideAtlas, MassIVE has already began to combine initially submitted and re-analyzed outcomes. Better help for submitted quantification outcomes can also be on the checklist of priorities. At current, the information containing quantitative data are made obtainable to obtain, however can’t be correctly built-in within the assets. One attainable answer is to realize mzTab help by the most well-liked proteomics evaluation instruments.