Following start, the breast-fed infant gastrointestinal tract is quickly colonized by a microbial consortium usually dominated by bifidobacteria. Accordingly, the full genome sequence of Bifidobacterium longum subsp. infantis ATCC15697 displays a aggressive nutrient-utilization technique concentrating on milk-borne molecules which lack a nutritive worth to the neonate.
Several chromosomal loci replicate potential adaptation to the infant host together with a 43 kbp cluster encoding catabolic genes, extracellular solute binding proteins and permeases predicted to be energetic on milk oligosaccharides. An examination of in vivo metabolism has detected the hallmarks of milk oligosaccharide utilization through the central fermentative pathway utilizing metabolomic and proteomic approaches. Finally, conservation of gene clusters in a number of isolates corroborates the genomic mechanism underlying milk utilization for this infant-associated phylotype.
PlasmoDB (http://PlasmoDB.org) is the official database of the Plasmodium falciparum genome sequencing consortium. This useful resource incorporates the not too long ago accomplished P. falciparum genome sequence and annotation, in addition to draft sequence and annotation rising from different Plasmodium sequencing tasks.
PlasmoDB at present homes data from 5 parasite species and gives instruments for intra- and inter-species comparisons. Sequence data is built-in with different genomic-scale knowledge rising from the Plasmodium analysis neighborhood, together with gene expression evaluation from EST, SAGE and microarray tasks and proteomics research. The relational schema used to construct PlasmoDB, GUS (Genomics Unified Schema) employs a extremely structured format to accommodate the various knowledge sorts generated by sequence and expression tasks.
A range of instruments permit researchers to formulate complicated, biologically-based, queries of the database. A stand-alone model of the database can also be obtainable on CD-ROM (P. falciparum GenePlot), facilitating entry to the knowledge in conditions the place web entry is troublesome (e.g. by malaria researchers working in the area). The objective of PlasmoDB is to facilitate utilization of the huge portions of genomic-scale knowledge produced by the international malaria analysis neighborhood. The software program used to develop PlasmoDB has been used to create a second Apicomplexan parasite genome database, ToxoDB (http://ToxoDB.org).
The Proteomics Identifications database: 2010 replace.
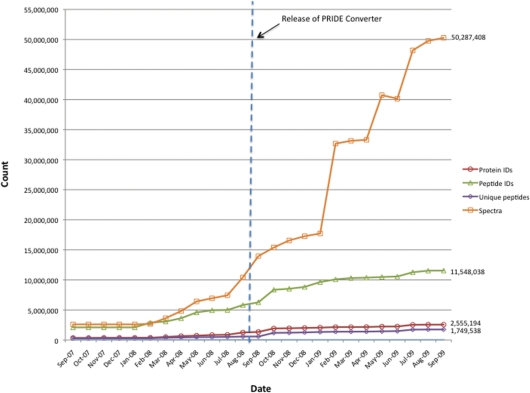
The Proteomics Identifications database (PRIDE, http://www.ebi.ac.uk/pride) at the European Bioinformatics Institute has turn out to be one of the principal repositories of mass spectrometry-derived proteomics knowledge. For the final 2 years, PRIDE knowledge holdings have grown considerably, comprising 60 completely different species, greater than 2.5 million protein identifications, 11.5 million peptides and over 50 million spectra by September 2009.
We right here describe a number of new and improved options in PRIDE, together with the revised submission course of, which now consists of direct submission of fragment ion annotations. Correspondingly, it’s now doable to visualise spectrum fragmentation annotations on tandem mass spectra, a key function for compliance with journal knowledge submission necessities.
We additionally describe current developments in the PRIDE BioMart interface, which now permits integrative queries that may be part of PRIDE knowledge to a rising quantity of organic assets similar to Reactome, Ensembl, InterPro and UniProt.
This means to carry out extraordinarily highly effective across-domain queries will definitely be a cornerstone of future bioinformatics analyses. Finally, we spotlight the significance of knowledge sharing in the proteomics area, and the corresponding integration of PRIDE with different databases in the ProteomExchange consortium.
The PRoteomics IDEntifications (PRIDE) database (https://www.ebi.ac.uk/pride/) is the world’s largest knowledge repository of mass spectrometry-based proteomics knowledge, and is one of the founding members of the international ProteomeXchange (PX) consortium. In this manuscript, we summarize the developments in PRIDE assets and associated instruments since the earlier replace manuscript was printed in Nucleic Acids Research in 2016. In the final three years, public knowledge sharing by way of PRIDE (as half of PX) has positively turn out to be the norm in the area. In parallel, knowledge re-use of public proteomics knowledge has elevated enormously, with a number of purposes.
We first describe the new structure of PRIDE Archive, the archival element of PRIDE. PRIDE Archive and the associated knowledge submission framework have been additional developed to assist the improve in submitted knowledge volumes and extra knowledge sorts. A brand new scalable and fault tolerant storage backend, Application Programming Interface and net interface have been applied, as a component of an ongoing course of. Additionally, we emphasize the improved assist for quantitative proteomics knowledge by way of the mzTab format. At final, we define key statistics on the present knowledge contents and quantity of downloads, and the way PRIDE knowledge are beginning to be disseminated to added-value assets together with Ensembl, UniProt and Expression Atlas.
A information to the Proteomics Identifications Database proteomics knowledge repository
The Proteomics Identifications Database (PRIDE, www.ebi.ac.uk/delight) is one of the principal repositories of MS derived proteomics knowledge. Here, we level out the principal functionalities of PRIDE each as a submission repository and as a supply for proteomics knowledge. We describe the principal options for knowledge retrieval and visualization obtainable by way of the PRIDE net and BioMart interfaces.
We additionally spotlight the mechanism by which tailor-made queries in the BioMart can be part of PRIDE to different assets similar to Reactome, Ensembl or UniProt to execute extraordinarily highly effective across-domain queries. We then current the newest enhancements in the PRIDE submission course of, utilizing the new easy-to-use, platform-independent graphical person interface submission software PRIDE Converter. Finally, we talk about future plans and the function of PRIDE in the ProteomExchange consortium.
In this chapter we describe the core Protein Production Platform of the Northeast Structural Genomics Consortium (NESG) and description the methods used for producing high-quality protein samples utilizing Escherichia coli host vectors. The platform is centered on 6X-His affinity-tagged protein constructs, permitting for an identical purification process for most targets, and the implementation of high-throughput parallel strategies.
In most instances, these affinity-purified proteins are sufficiently homogeneous {that a} single subsequent gel filtration chromatography step is sufficient to provide protein preparations which can be larger than 98% pure. Using this platform, over 1000 completely different proteins have been cloned, expressed, and purified in tens of milligram portions over the final 36-month interval (see Summary Statistics for All Targets, ).
Our expertise utilizing a hierarchical multiplex expression and purification technique, additionally described on this chapter, has allowed us to attain success in producing not solely protein samples but in addition many three-dimensional buildings.
As of December 2004, the NESG Consortium has deposited over 145 new protein buildings to the Protein Data Bank (PDB); about two-thirds of these protein samples had been produced by the NESG Protein Production Facility described right here. The strategies described right here have confirmed efficient in producing high quality samples of each eukaryotic and prokaryotic proteins. These improved robotic and?or parallel cloning, expression, protein manufacturing, and biophysical screening applied sciences can be of broad worth to the structural biology, useful proteomics, and structural genomics communities.