The PRoteomics IDEntifications (PRIDE, http://www.ebi.ac.uk/pride) database on the European Bioinformatics Institute is among the most distinguished information repositories of mass spectrometry (MS)-based proteomics information. Here, we summarize latest developments in the PRIDE database and associated instruments. First, we offer up-to-date statistics in information content material, splitting the figures by teams of organisms and species, together with peptide and protein identifications, and post-translational modifications.
We then describe the instruments which can be a part of the PRIDE submission pipeline, particularly the not too long ago developed PRIDE Converter 2 (new submission device) and PRIDE Inspector (visualization and evaluation device). We additionally give an replace concerning the integration of PRIDE with different MS proteomics assets in the context of the ProteomeXchange consortium. Finally, we briefly overview the standard management efforts which can be ongoing at current and define our future plans.
The PRoteomics IDEntifications database on the European Bioinformatics Institute is among the most distinguished information repositories of mass spectrometry (MS)-based proteomics information. Here, we summarize latest developments in the PRIDE database and associated instruments. First, we offer up-to-date statistics in information content material, splitting the figures by teams of organisms and species, together with peptide and protein identifications, and post-translational modifications. We then describe the instruments which can be a part of the PRIDE submission pipeline, particularly the not too long ago developed PRIDE Converter 2 (new submission device) and PRIDE Inspector (visualization and evaluation device). We additionally give an replace concerning the integration of PRIDE with different MS proteomics assets in the context of the ProteomeXchange consortium. Finally, we briefly overview the standard management efforts which can be ongoing at current and define our future plans.
INTRODUCTION
Mass spectrometry (MS)-based proteomics approaches are extensively used in the life sciences. There are three foremost workflows, with bottom-up proteomics essentially the most extensively used approach (often known as shot-gun proteomics) . In this experimental arrange, the proteins to be analysed are enzymatically digested by a protease (most frequently trypsin) into doubtlessly extremely complicated peptide mixtures, that are then subjected to fractionation by multidimensional liquid chromatography steps earlier than they’re measured in the mass spectrometer. Other foremost approaches are top-down, the place intact proteins are measured and focused proteomics (corresponding to Selected Reaction Monitoring, SRM), the place the researcher tries to detect particular proteins in a given pattern .
The PRoteomics IDEntifications database was initially arrange in 2004 ( to allow public information deposition in the MS proteomics discipline, and to help the experimental information described in publications in the course of the manuscript overview course of. The foremost information sorts saved in PRIDE are protein and peptide identifications (IDs) and quantitative values (together with post-translational modifications, PTMs), the analysed mass spectra and the associated technical and organic metadata. PRIDE helps bottom-up proteomics approaches, primarily tandem MS (MS/MS) information, but additionally Peptide Mass Fingerprinting datasets, and presents the information as initially analysed by the researchers, with a number of common search engines like google and yahoo/evaluation workflows totally supported.
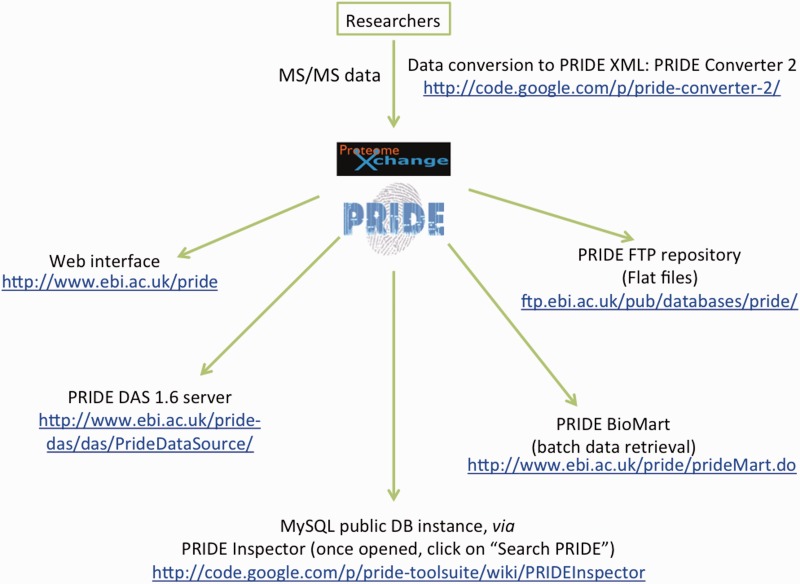
Unlike different MS proteomics assets, corresponding to PeptideAtlas ) and the Global Proteome Machine Database (GPMDB) ( no reprocessing of the information is carried out as a result of PRIDE goals to mirror the writer’s evaluation view on the experimental information. In reality, PRIDE stays because the distinctive generic useful resource of this sort since National Center for Biotechnology Information (NCBI) the repository Peptidome its sibling useful resource in the USA, was discontinued in April 2011. Other MS information repositories, corresponding to MaxQB , are extra specialised [for an extensive review, see (or are restricted to 1 explicit evaluation workflow.
For SRM information, the brand new PeptideAtlaS SRM Experiment Library (PASSEL) ( is the principle accessible useful resource. At current, there is no such thing as a extensively used useful resource dedicated to top-down proteomics approaches. In addition to the ‘pure’ MS proteomics assets, there are different databases that may current an additional layer of knowledge on prime of the MS experiments with out storing the underlying mass spectra. Some not too long ago developed databases of this sort are the Model Organism Protein Expression Database (MOPED) ), PaxDB (each of them centered on protein expression data) and neXtProt ).
Several providers have been developed by the PRIDE staff, that are closely utilized by exterior customers but additionally by PRIDE itself, particularly the ‘Protein Identifier Cross-Reference’ (PICR) service (a protein identifier mapping useful resource) and the ‘Ontology Lookup Service’ (OLS) (to question, browse and navigate biomedical ontologies) (. In addition, ‘Database on Demand’ is a service to generate tailor-made databases for performing proteomics searches .
As a key level, to enhance and make the information submission course of simpler, a number of instruments have additionally been made accessible to the proteomics group, corresponding to the favored PRIDE Converter , PRIDE Inspector and the brand new PRIDE Converter 2 (. It is essential to focus on that every one the softwares, together with the PRIDE core and internet modules are developed in Java and are open supply. PRIDE is a really useful submission website of key journals corresponding to Proteomics, Molecular and Cellular Proteomics and Nature Biotechnology. Currently, scientific journals and funding companies alike are more and more mandating public deposition of MS information to help the publication of associated proteomics manuscripts.
In this manuscript, we summarize developments in the PRIDE database and associated instruments for the reason that earlier Nucleic Acids Research (NAR) database replace We may even define the PRIDE information deposition course of, introduce the ProteomeXchange (PX) consortium and high quality management (QC) efforts and spotlight future developments.
DATA CONTENT IN PRIDE AND HOW TO ACCESS IT
There has been a considerable enhance in the quantity of saved information in PRIDE in the course of the previous years. By September 2012, PRIDE incorporates 25 853 MS-based proteomics experiments (in contrast with 9908 when the final NAR manuscript was submitted, in September 2009), round 11.1 million recognized proteins (2.5 million in September 2009), 61.9 million recognized peptides (11.5 million in September 2009) and 324 million spectra (50.Three million in September 2009). Note that these information holdings are absolute figures, not distinguishing public and pre-publication information. At the second of writing, 66.7% (17 219) of the experiments have been publicly accessible.
The full set of information in PRIDE includes 323 taxonomy identifiers (in contrast with 60, in September 2009), together with human and many mannequin organisms nonetheless present nearly all of the information. A complete of 89 animal species are represented, contributing 62.2% and 61.9% of all protein and peptide IDs in PRIDE, respectively. Human continues to be essentially the most represented species (28.1% and 37.2% of protein and peptide IDs, respectively). As a matter of reality, human and mouse alone account for nearly as many IDs as the opposite species collectively: 44.2 % and 49.4 % of the protein and peptide IDs, respectively.
However, the relative proportion of different teams of organisms has elevated, particularly in the case of vegetation (46 taxonomy identifiers, 19.3% and 14.8%, respectively) and micro organism (12.7% and 17.4%, respectively). Bacteria are once more by far the group of organisms with the very best variety of taxonomy identifiers (122).
Fungi are additionally represented (22 taxonomy identifiers, 2.7 and 3.0%, respectively). Apart from human, essentially the most represented organisms in PRIDE are (in this order) mouse, maize, Arabidopsis, Bacillus subtilis, pig, rat, Drosophila, zebrafish, Puniceispirillum marinum, Escherichia coli and Saccharomyces cerevisiae.
consists of essentially the most plentiful PTMs current in the database. Not surprisingly, essentially the most usually discovered PTM is oxidation (5.7 million modified websites), primarily as a result of excessive quantity of methionine oxidation, a modification that may be biologically related
PRIDE Inspector
In addition, by means of the ‘Search PRIDE Database’ possibility, PRIDE Inspector can entry information already in PRIDE for information mining functions utilizing the PRIDE public MySQL occasion, which is up to date repeatedly. Apart from being a stand-alone device, as talked about earlier than, PRIDE Inspector may also be accessed utilizing Java Web Start on the PRIDE internet web page.
PRIDE and the ProteomeXchange consortium
A significant focus of PRIDE improvement in the previous 2 years was to make sure on the very least minimal annotation of experiments and to carry out fundamental high quality checks of the submitted information to PRIDE. The improvement of PRIDE Inspector was the important thing step ahead in that course, and lots of its elements have been used in the inner PRIDE submission pipeline. The automated pipeline permits the detection of clear errors in the submitted information which can be notified to the submitter and can then be corrected Finally, the event of the PRIDE Converter 2 as the brand new submission device has improved consistency on the degree of experimental annotation.
In 2013, we are going to finalize and launch a brand new useful resource referred to as PRIDE-Q, as a quality-controlled subset of PRIDE, which may fulfil each a minimal degree of annotation and Peptide Spectrum Match high quality requirements.
DISCUSSION
In the previous Three years, PRIDE has labored on two foremost duties: (i) improvement of strong information submission pipelines (corresponding to the event of the PRIDE Converter 2), together with the preliminary implementation of the PX consortium information workflow, and the chance to seize quantification data in a standardized manner; and (ii) institution of QC checks, together with the event of PRIDE Inspector and an inner information submission pipeline, in a position to flag apparent errors that may be then communicated to the submitters.
However, many extra future efforts will probably be wanted in each instructions. We are engaged on the event of the brand new PRIDE system (together with a brand new database schema and internet interface) that can totally help the PSI requirements mzML and mzIdentML. This will probably be a gradual course of, and help for these codecs will probably be added sequentially to the PRIDE system (additionally as submission codecs) and instruments, whereas we may even preserve supporting PRIDE XML.
On the opposite hand, the discharge of PRIDE-Q, envisioned to assist non-expert proteomics biologists to ‘digest’ the possibly complicated data coming from MS information, will occur in 2013. However, the standard necessities will have to be refined and will evolve dynamically over time.
It is value highlighting that PRIDE has been used for analysis functions in a number of latest research involving the meta-analysis of mixed information coming from very totally different proteomics experimental setups , the development and evaluation of current protein sequence databases ) or to help genomics-related findings ( Some analysis has additionally been carried out to reveal the usefulness of information in PRIDE to carry out a posteriori QC of the saved information (). We count on that this pattern will proceed to develop in the close to future, and that PRIDE continues on a trajectory from a publication-centric repository to an integrative useful resource for MS-based protein expression information.
PRIDE will preserve enjoying an essential function for the group, additionally in the context of the nascent Human Proteome Project . We invite events in PRIDE developments (together with the associated software program and instruments) to comply with the PRIDE Twitter account (@pride_ebi).